Purpose:
The purpose of this lab was to study transformation and the effect that integrating certain genes into a typical E. Coli bacteria would have on the cell. This lab also explored the effect that certain environments would have on the bacteria, including those containing antibiotics or certain sugar molecules, as well as how the introduced gene would interact with these environments.
Introduction:
Genetic transformation is a change caused by the introduction of a gene, and how it is incorporated into a cell’s DNA sequence. Once this particular gene is added into the DNA, it can code for related mRNAs that code for particular proteins that give the cell different properties than it had before. This transformation can be helpful or harmful, depending on the DNA sequence introduced into the cell.
Methods:
To begin with, we labeled one closed micro test tube +pGLO and another -pGLO, to indicate which E. Coli bacteria would be given the chance to incorporate the gene into its DNA.
+pGLO tube
-pGLO tube
Afterwards, 20 microliters of transformation solution was transferred into each tube using a sterile transfer pipet, segmented into 10 microliter segments. With the transformation solution in hand, the tubes were iced.
Ice to meet you, micro test tubes...
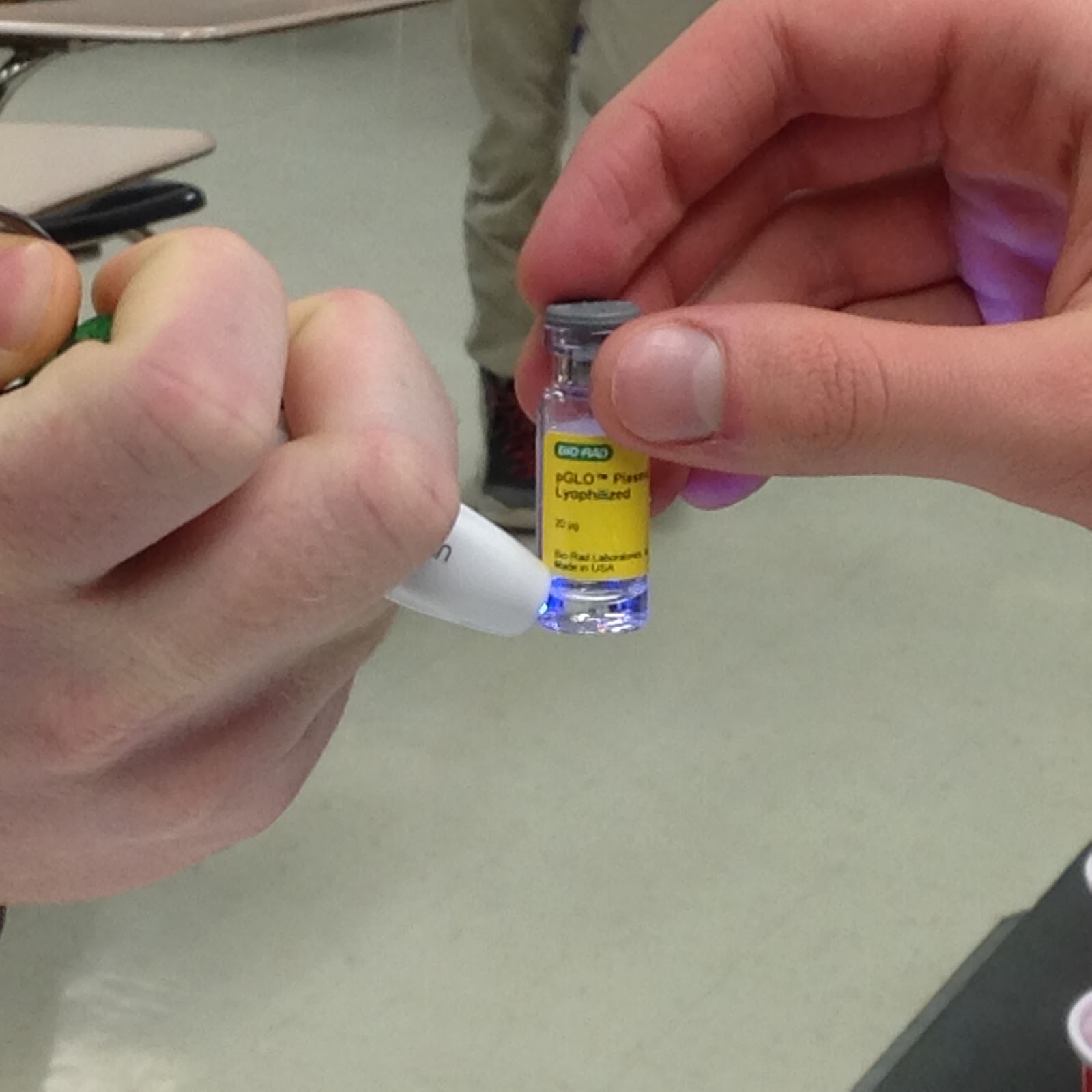
Next, we used a sterile loop to transfer a colony of E. Coli bacteria into each of the test tubes. The loop was spun thoroughly to fully incorporate the colony into each of the tubes. Afterwards, we used a UV light to determine that E. Coli was unable to glow on its own. Just after this was done, we used a different loop to incorporate pGLO plasmid DNA into the +pGLO tube, and noted that the solution (but not the bacteria) in the +pGLO tube glowed under UV light, but the -pGLO tube did not (as it should!).
Our reports show that the tube without any pGLO DNA does not, in fact, glow…
But the tube with the pGLO does. Shocking!
Also, note that the pGLO does glow on its own. So there’s no eldritch magicks at play here.
Hard to tell, I know, but this solution of pGLO DNA is glowing.
Once this was completed, it was back to the ice for ten minutes.
Ice day today, isn’t it?
Meanwhile, we gathered the LB nutrient agar plates that we would use to culture more bacteria. They were already pre-labeled and pre-made, so we didn’t really need to worry about differentiating them.
Oh hi plates.
Next came the heat shock! We removed the tubes from the ice and put them in 42 degrees Celsius water for 50 seconds.
HEAT SHOCK
After that, we put the tubes back on ice for two minutes to seal the E. Coli’s breached membranes with, hopefully, the pGLO integrated into the ones in the +pGLO tube. Once that was completed, we added 250 microliters of nutrient broth to the tubes, closed them, and mixed the solutions together by tapping the side of the tube. We then incubated them for ten minutes at room temperature.
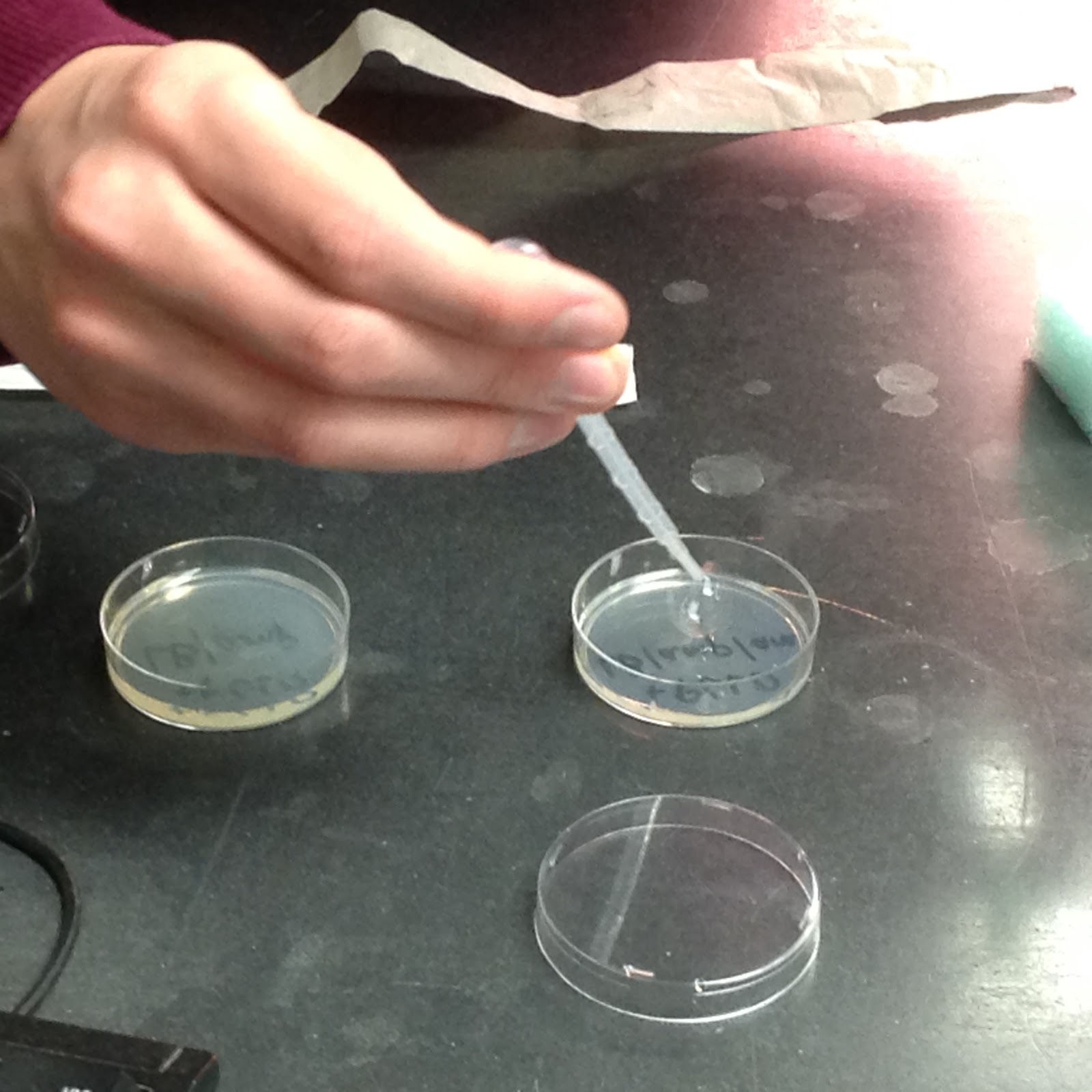
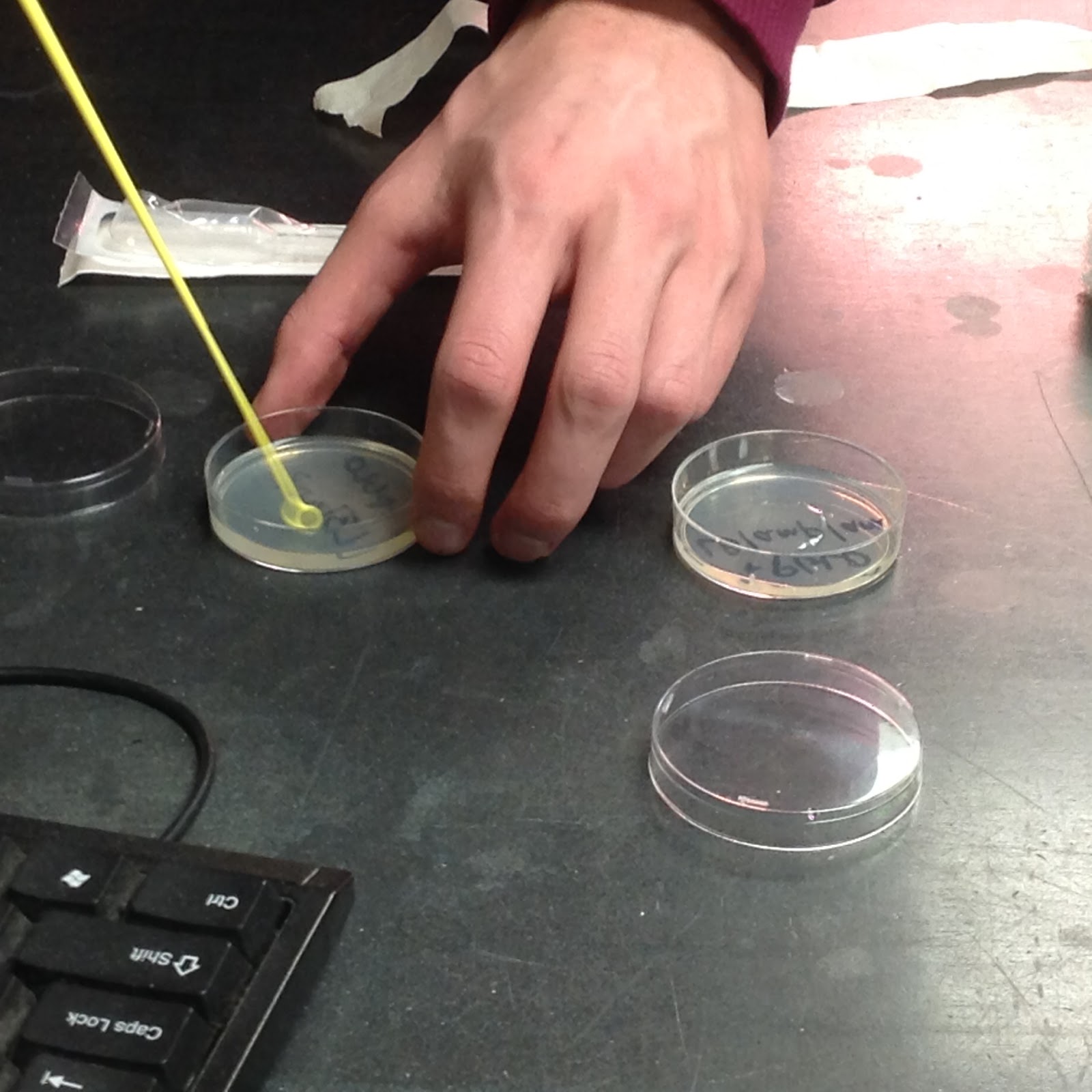
All that was left after that was to pipet 100 microliters of each solution onto their respective plates. The +pGLO tube was pipetted onto the +pGLO plates, one with LB and amp and the other with LB, amp, and ara. The -pGLO tube was pipetted onto the -pGLO plates, one with LB/amp and the other just with LB. Using a sterile loop for each plate, we spread the solutions along the surface of each plate to fully disperse the bacteria into their new home.
The process of integrated the bacteria into their new environments
Once that was done, we stacked up the plates upside down and incubated them for a day to allow the bacteria to grow.
Topsy-turvy
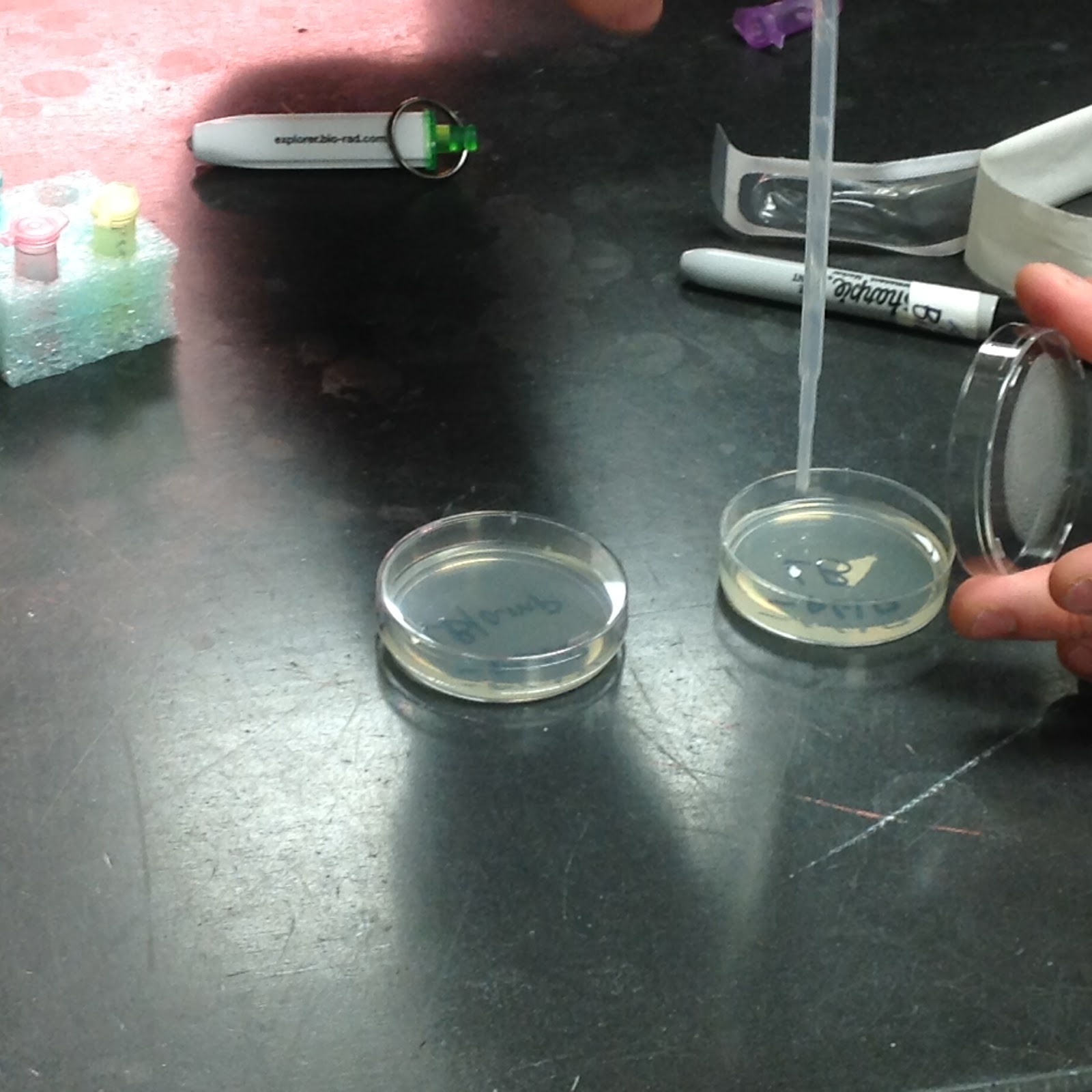
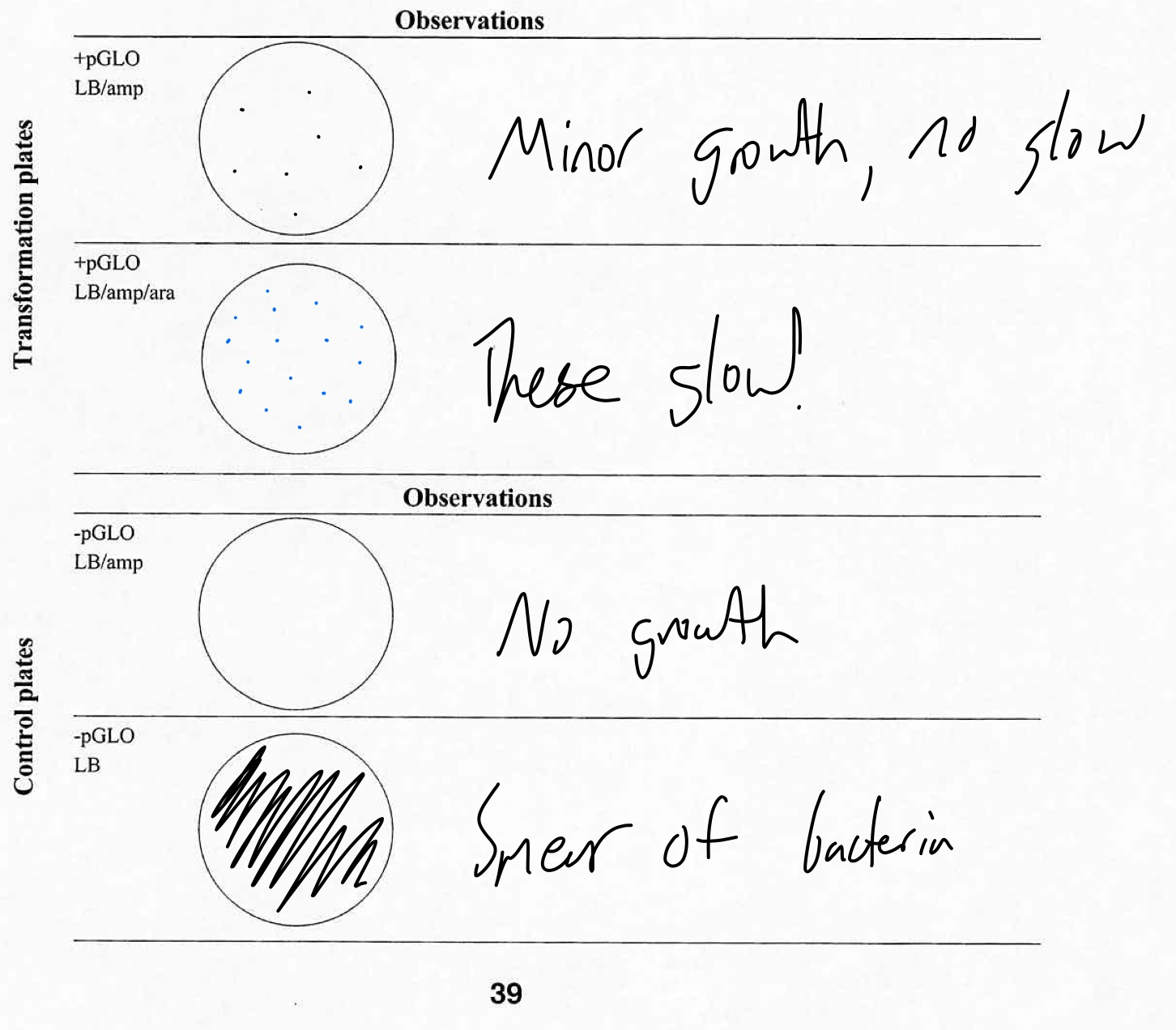
The next day, we reclaimed our cultures and opened them all up to examine them. While my groupmate responsible for the pictures did not include the first three plates, he did include the final one, being the +pGLO, LB/amp/ara plate. This culture managed to grow in spite of proximity to the ampicillin, which is normally an E. Coli killer. It also glows, too. Interesting!
Straight Outta Chipotle. Now with built-in rave lighting!
While we have no picture-proof for the other three cultures, I will describe them here. The -pGLO plate with only the LB was identical to the E. Coli culture we obtained the bacteria from the prior day, spread about the culture in many smears. The -pGLO plate with the ampicillin included had no growth, as the bacteria had no protection from the ampicillin and could not sustain itself. The +pGLO plate with the ampicillin did show growth, since the pGLO gene protects the bacteria from the antibiotic, but did not glow as no arabinose was available to allow the gene to function.
Once the lab was done, we killed all remaining E. Coli with bleach and disposed of all materials that the E. Coli inhabited. We wouldn’t want PHS to become the next Chipotle, now would we?
Data:
As stated in the purpose, the -pGLO plate with only the LB was identical to the E. Coli culture we obtained the bacteria from the prior day, spread about the culture in many smears. The -pGLO plate with the ampicillin included had no growth, as the bacteria had no protection from the ampicillin and could not sustain itself. The +pGLO plate with the ampicillin did show growth, since the pGLO gene protects the bacteria from the antibiotic, but did not glow as no arabinose was available to allow the gene to function. And the +pGLO plate with ampicillin and arabinose showed growth, thanks to the pGLO gene protecting the bacteria from the antibiotic, and was able to glow since arabinose was available for the gene to function.
Graphs and Charts:
pGLO plate observation chart
Discussion:
The E. Coli showed several changes when transformed by the pGLO plasmid DNA. The bacteria that incorporated the pGLO DNA into its own DNA was able to grow in conditions with ampicillin present, and glow under UV light. Bacteria without this gene had no natural fluorescence and could not survive in ampicillin-rich environments, as indicated by the above table.
The arabinose sugar plays a role in the ability for the pGLO’s glowing part of the gene to function. Without arabinose in the environment, it cannot naturally glow, as indicated by the +pGLO LB/amp plate.
We know for a fact that pGLO is responsible for these changes because the original bacteria did not glow naturally (as we determined during the procedure) and because E. Coli without the pGLO were not able to survive in ampicillin. And we know that this is not a naturally occurring trait within E. Coli because only the bacteria exposed to the plasmid were able to display these features, proving that transformation is required for such an effect to take place. We know that transformation definitely occurred in the +pGLO tube.
E. Coli was able to grow on the LB plate that did not contain any ampicillin or arabinose. Quite a lot, in fact! By looking at them, however, it is not fully determinable (though it is easy to hypothesize) whether or not the bacteria are ampicillin resistant. If we look at the plate that contained the ampicillin, we see no growth, and since both shared in the fact that they have no pGLO gene, it is easy to postulate that the bacteria on the LB only plate are not antibiotic resistant.
For the green glow to exist, arabinose must be present, UV light must be shining upon the cultures, and the pGLO gene must be incorporated into the cell’s DNA. Only the plate that was +pGLO and had arabinose available was able to grow, and it only glowed when exposed to UV light. UV light must be eliciting some sort of response within the bacteria cells, forcing the gene to activate and be transcribed to produce whatever chemical makes the cell grow. An advantage that turning genes on and off at will has is the ability for cells to only activate a vital gene when it is necessary. In the bacteria’s case, the bacteria only needs to glow its green color when UV light is present, and the cells capture and use the UV energy to glow without expending any of its own resources.
Conclusion:
Transformation is a vital aspect of genetics to understand because incorporating segments of DNA into other organisms, including humans, is extremely useful. If there is a deadly disease or mutation that can be mitigated through incorporating a certain segment of plasmid DNA, it is simple to do in the zygotic stage of an organism’s development. When the cell divides, so too does the gene, and it spreads throughout the whole of the body to better the organism’s life.
References:
Unless otherwise stated, all information related to transformation has been synthesized from Jane Reece’s Campbell Biology AP Edition Textbook and the pGLO Transformation Student Manual.
Paraphrased procedures have been synthesized from procedures found in the pGLO Transformation Student Manual.
The pGLO plate observation chart was lifted from the pGLO Transformation Student Manual, but all data is original to our experiences.
- All pictures are original from the in-class experiment.